





| Research Article | ||
J Microbiol Infect Dis. 2023; 13(4): 182-189 J. Microbiol. Infect. Dis., (2023), Vol. 13(4): 182–189 Original Article In silico analysis, modeling, and docking studies on the EtfB and EtfC virulence proteins of the fish pathogen Edwardsiella tardaBalakrishnan Naveen Rajeshwar1* and Thangapalam Jawahar Abraham21Department of Aquatic Animal Health Management, ICAR-Central Institute of Fisheries Education, Mumbai, India 2Department of Aquatic Animal Health, Faculty of Fishery Sciences, West Bengal University of Animal and Fishery Sciences, Kolkata, India *Corresponding Author: B. Naveen Rajeshwar. Present address: Department of Aquatic Animal Health Management, ICAR-Central Institute of, Fisheries Education, Mumbai, India. Email: balkirajeshwar [at] gmail.com Submitted: 13/08/2023 Accepted: 14/12/2023 Published: 31/12/2023 © 2023 Journal of Microbiology and Infectious Diseases
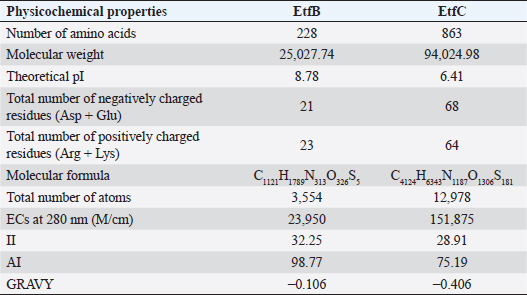
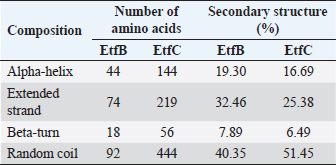
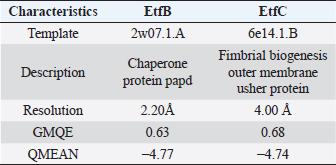
AbstractBackground: In-silico analysis and molecular docking tools offer alternative ploy to characterize the virulence factors of Edwardsiella tarda and identify potential herbal antimicrobial compounds as substitutes to overcome the adverse health effects of antibiotics. Aim: In this study, fimbrial adhesion proteins of E. tarda namely EtfB and EtfC were regarded as virulence factors for bioinformatic analysis. Methods: The virulent protein sequences, recovered from the UniProt database, were analyzed structurally and functionally. SWISS-MODEL generated the homology models of these proteins and verified the quality. The predicted 3D models were used as receptors individually in docking studies against select herbal compounds as ligands for these proteins. Results: Both proteins were soluble, hydrophobic, and thermally stable. The predicted models’ Z-values were within the scores particularly found for native proteins of the same size, indicating that the models were suitable. The molecular docking identified corilagin (Terminalia chebula), lutein (Amaranthus viridis), isoginkgetin (Cyperus rotundus), amentoflavone (C. rotundus), and acacic acid (Acacia concinna) as the best herbal compounds against EtfB gene, and sciadopitysin (C. rotundus), oleandrin (Nerium oleander), isoginkgetin (C. rotundus), ginkgetin (C. rotundus), and cyanidin 3-sophoroside (Hibiscus rosa-sinensis) against EtfC gene based on the lowest E-values in order. Conclusion: This study revealed that the fimbrial proteins are potential drug targets. Docking results predicted that these low E-value herbal compounds may control edwardsiellosis in aquaculture, which can be further validated using in vitro and in vivo studies. The bioinformatics tools save time, cost, and energy in finding promising drugs against a specific target of interest. Keywords: Edwardsiellosis, Edwardsiella tarda, Virulence proteins, Herbal compounds, Molecular docking. IntroductionThe aquaculture sector contributes significantly to the economy yet is adversely affected due to frequent diseases, mostly due to intensive cultural practices for higher economic gain (Mishra et al., 2017). It is estimated that the diseases account for 5%–10% of the production cost (Mishra et al., 2017). Edwardsiellosis, caused by Edwardsiella spp. from the Enterobacteriaceae family, is accountable for major global losses in important wild and cultured fish species (Sakai et al., 2007; Reichley et al., 2017). Edwardsiella tarda is the best-studied species in the Edwardsiella genus and is a versatile pathogen with a vast variety of hosts, including fish. This pathogen causes disease in reptiles, birds, humans, and other mammals (Fogelson et al., 2016). Antibiotic treatment of bacterial infections has been compromised by the emergence of antibiotic resistance (Watts et al., 2017). Alternative eco-friendly approaches without side effects have been advocated in disease control strategies. Herbal compounds with specific antimicrobial properties are among the best alternatives for use as dietary supplements (Shakya, 2017). In this regard, bioinformatics plays a vital role in verifying the herbal compounds’ efficacy against a pathogen. It bridges a disease in the field with drug discovery in the laboratory. Edwardsiella tarda possesses essential virulence factors that increase the chance of bacterial survival and pathogenesis in hosts (Park et al., 2012), which is not entirely understood yet believed to be a multifactorial process. Edwardsiella tarda is involved in the secretion of adhesions, degradative enzymes, type III secretion system, and type VI secretion system. The commonly identified virulence genes in E. tarda isolates are catalase (katB), TTSS regulator (esrB), putative killing factor (mukF), fimbrial operon (fimA), glutamate decarboxylase (gadB), citrate lyase ligase (citC), phosphate specific transport (pstS, pstC), astA, isor, outer membrane protein(OMP) (ompS2), transcriptional regulator (ssrB), hemolysin A (hlyA), and ATPase domain of DNA Gyrase (gyrB) (Moustafa et al., 2016). Jin et al. (2012) investigated the role of NanA, a sialidase, in the pathogenesis of E. tarda in Japanese flounder. The essential genes of OMPs of the fish pathogen E. tarda were identified, characterized, and analyzed, which could be apt as a vaccine or drug candidate (Neema et al., 2011). The modeled novel OMP of E. tarda was docked with the selected antibiotics to assess the potential drug doxycycline to combat edwardsiellosis (Devi and Jo, 2015). The main challenge in commercial aquaculture is to identify and characterize the suitable drug target leading to the discovery of herbal drugs as alternatives to antibiotics. A better understanding of the interactions between virulence factors will lead to the development of novel antimicrobials against E. tarda (Leung et al., 2012). Since the adhesion of infective bacteria to host surfaces is a crucial step in dissemination, virulence factors involved in host attachment are considered objects of study. The fimbriae or fimbrial gene cluster of E. tarda seemed responsible for adhesion with the host. The fimbrial genes such as etfA, etfB, etfC, and etfD constitute the E. tarda fimbrial gene cluster and are used to distinguish between fish infective and noninfective strains of E. tarda (Sakai et al., 2007). The virulent E. tarda strains get attached to the host surface using fimbriae. The fimbrial Etf proteins are the parts of the FimA protein involved in adhesion. They play a crucial role in the entry of pathogens into the host using various mechanisms leading to disease occurrence. Many potential virulence factors associated with fimbriae that cause E. tarda infection are identified. In this study, in silico analysis of E. tarda fimbrial chaperon protein (EtfB) and fimbrial usher protein (EtfC) was conducted to highlight their physicochemical, functional, and secondary structural properties and homology modeling of proteins. The modeled proteins are considered drug targets as fimbriae are an essential factor in E. tarda virulence. They were docked with herbal compounds to identify the effective herbal drug targets to control E. tarda infection. Material and MethodsSequence retrievalThe amino acid sequences of the E. tarda fimbrial chaperon protein (EtfB) and fimbrial usher protein (EtfC) with accession numbers Q84IF7 and Q7DFZ6, respectively, were recovered from the Universal Protein Resource (UniProt) [https://www.uniprot.org/]. It was obtained in FASTA format and used in additional structural and functional analyses. Prediction of physicochemical propertiesUsing the ExPASy-ProtParam prediction server [https://web.expasy.org/protparam/], the physicochemical characteristics such as amino acid composition, the number of amino acids, molecular weight, extinction coefficient (EC), aliphatic index (AI), instability index (II), grand average hydropathy (GRAVY), isoelectric point (pI), molecular formula, total number of atoms, and number of positively and negatively charged residues of EtfB and EtfC proteins were determined. Functional analysisThe SOSUI server identified the protein’s nature (http://harrier.nagahama-i-bio.ac.jp/sosui/sosui_submit.html). The CYS_REC tool estimated the presence or absence of disulfide bonds that determine the functional linkage and protein stability, their bonding pattern, and the number of cysteine residues (http://linux1.softberry.com). Secondary structure characterizationSOPMA (self-optimized prediction method with alignment) tool was employed to acquire the secondary structures of EtfB and EtfC proteins such as alpha-helix, extended strand, beta-turn, and the random coil [https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html]. Homology modeling and evaluation of the tertiary structureUsing the template structure from the protein structural database (PDB), a completely automated protein structural homology-modeling server (SWISS-MODEL server) carried out the homology modeling of the 3D structure of EtfB and EtfC proteins. It was accessed using the Expasy web server (https://swissmodel.expasy.org/), which identified the structural template and aligned the target sequence with the template structure. With the help of a template, it built the models and analyzed their quality. The qualities of the predicted models were analyzed by RAMPAGE (Lovell et al., 2003), ProQ (Cristobal et al., 2001), ProSA-web (Wiederstein and Sippl, 2007), and Ramachandran plot (Ramachandran et al., 1963). Molecular dockingThe virulent fimbrial EtfB and EtfC proteins were regarded as drug targets because fimbriae play a vital role in host adhesion. The potent herbal compounds from various sources act like ligands to cling to virulent fimbrial proteins. The chemical database retrieved their structures out of PubChem (https://pubchem.ncbi.nlm.nih.gov/). The 3D chemical structures have been downloaded in structure data file (SDF) format. Since molecular docking requires the target and ligand in the form of a PDB file, Open Babel software was used for converting the ligand from SDF to PDB format. HEX 8.0.0.0 Cuda (Macindoe et al., 2010) and HDOCK server (Yan et al., 2020) performed docking studies on the receptor and ligand PDB structure. ResultsPhysicochemical properties predictionAs shown in Table 1, the fimbrial proteins EtfB and EtfC had 228 and 863 amino acids, respectively. The calculated pI value of the EtfB protein is 8.78 and the EtfC protein is 6.41. The total number of positive (Arg + Lys) of 21 and negative charge residues (Asp + Glu) were 21/23 for EtfB and 68/64 for eftC proteins. The ECs of EtfB and EtfC proteins measured at 280 nm were 23,950 and 151,875 M/cm, respectively. The II of the EtfB protein is 32.25, whereas the EtfC protein is 28.91. The AI was predicted to be 98.77 for EtfB and 75.19 for EtfC proteins. The GRAVY values of EtfB and EtfC proteins of E. tarda were −0.106 and −0.406, respectively. The amino acid composition of EtfB and EtfC proteins computed using ExPASyProtParam is given in Table 2. Functional analysisThe EtfB and EtfC proteins of E. tarda were soluble. Six cysteines were present in the EtfC and none in the EtfB. None of the cysteines formed a disulfide bond. Secondary structure characterizationAs revealed in Table 3, the alpha helix constituted 19.30% and 16.69% of the EtfB and EtfC proteins, respectively. The extended strand compositions of EtfB and EtfC proteins were 32.46% and 25.38%, respectively. About 40.35% and 51.45% of random coils were found in the EtfB and EtfC proteins, respectively. The beta turns accounted for 7.89% of EtfB and 6.49% of EtfC proteins. Homology modelingThe 3D models with the best sequence identity were built for EtfB and EtfC proteins of E. tarda using the SWISS-MODEL server (Fig. 1a and b). The EtfB protein was built with the template 2w07.1.A named chaperone protein papd at a resolution of 2.20 Å. The EtfC protein was built with the template 6e14.1.B named fimbrial biogenesis outer membrane usher protein at a resolution of 4.00 Å. The global model quality estimate (GMQE) scores of EtfB and EtfC proteins are 0.63 and 0.68, respectively. The QMEAN Z-scores were −4.77 for EtfB and −4.74 for EtfC proteins. Table 1. Physicochemical properties of the EtfB and EtfC proteins of E. tarda.
Table 2. Amino acid composition of the EtfB and EtfC proteins of E. tarda.
Table 3. Secondary structure elements of the EtfB and EtfC proteins of E. tarda computed using the SOPMA tool.
Table 4. Homology modeling data of the EtfB and EtfC proteins of E. tarda by SWISS-MODEL accessed via the ExPASy web server.
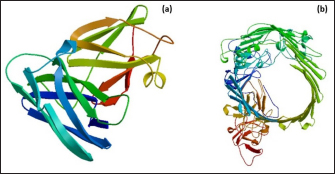
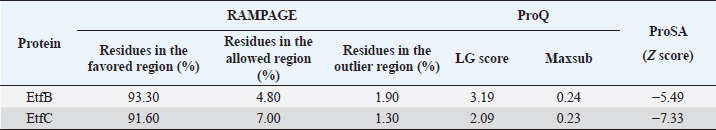
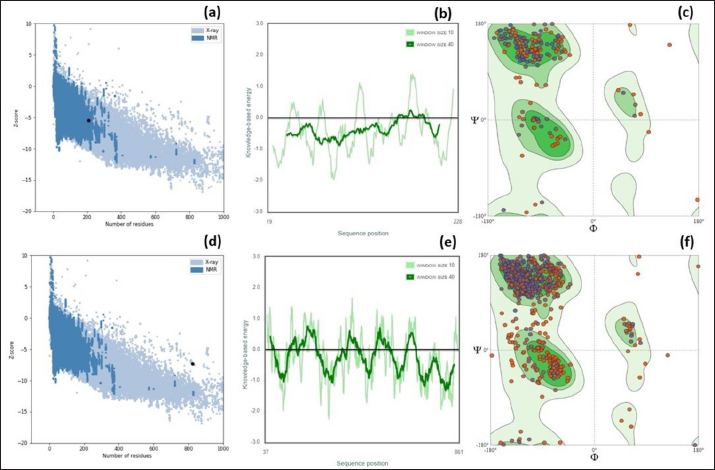
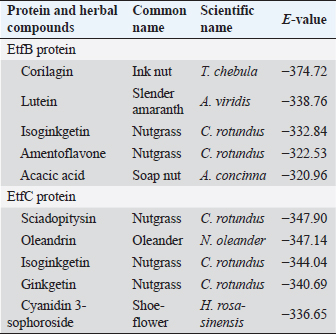
Fig. 1. Homology modeling of the (a) EtfB and (b) EtfC proteins of E. tarda by SWISS-MODEL, accessed via the ExPASy web server. Evaluation of tertiary structureIn RAMPAGE analysis, the residues in the favored region of EtfB and EtfC proteins of E. tarda were 93.3% and 91.6%, respectively (Table 5). The residues in the allowed region of EtfB and EtfC proteins were 4.8% and 7.0%, respectively, indicating improved model quality. The residues in the outlier region of EtfB and EtfC proteins of E. tarda were 1.9% and 1.3%, respectively. The ProQ validated the protein based on several structural features and predicted the protein model quality. The respective LG scores of EtfB and EtfC were 3.186 and 2.092. The MaxSub values were 0.238 and 0.232, respectively, for EtfB and EtfC. The results of the ProSA-web validation on the predicted models are represented in three forms (Table 5; Fig. 2a–d). The Z-scores of the ProSA web were given in the form of an image (Fig. 2a and b). The Z-scores of the predicted models of EtfB and EtfC proteins were −5.49 and −7.33, respectively (Fig. 2d and e). The ProSA-web server also gave the plot of residue scores. The Ramachandran plot analysis is shown in Figure 2c and f. Molecular dockingThe present study used the EtfB and EtfC proteins of E. tarda as receptors and asiatic acid, crucumin, allicin, corilagin, phyllanthin, nimbin, lupeol, campesterol, 6-gingerol, aloin, and so on, as main ligands for docking by HEX 8.0.0.0 Cuda software. The product with lower net binding free energy is thought to be the optimal ligand for the target protein. The docking results provided the least binding free energy (ΔGbind) or E-value for corilagin (−374.72; Ink nut–Terminalia chebula), lutein (−338.76; slender amaranth–Amaranthus viridis), isoginkgetin (−332.84; nutgrass–Cyperus rotundus), amentoflavone (−322.53; nutgrass–C. rotundus), and acacic acid (−320.96; Soap nut–Acacia concinna) in order against EtfB gene. As for the EtfC gene, sciadopitysin (−347.90; nutgrass–C. rotundus), oleandrin (−347.14; oleander–Nerium oleander), isoginkgetin (−344.04; nutgrass–C. rotundus), ginkgetin (−340.69; nutgrass–C. rotundus), and cyanidin 3-sophoroside (−336.65; shoe-flower–Hibiscus rosa-sinensis) gave the lowest E-values. The results were tabulated in Table 6. Molecular docking of top herbal antimicrobial compounds with virulence proteins was carried out using HDOCK software and pictorially represented with amino acid interactions in Figure 3. DiscussionThe pI value of more than seven indicated that EtfB protein is alkaline and less than seven indicated that EtfC protein is acidic in nature. The ECs of proteins are measured at 280 nm as the maximum absorbance of proteins in the UV spectra was caused by aromatic tryptophan and tyrosine residues and a small amount of phenylalanine (Lakowicz, 2013). These computed values may be helpful in quantitatively studying protein–protein and protein-ligand interaction in solution. The II of the EtfB (32.25) and EtfC (28.91) proteins of E. tarda were <40, and therefore, both proteins are considered stable (Guruprasad et al., 1990). The AI was higher for EtfB and EtfC proteins, thus indicating better thermal stability of these proteins (Juibari et al., 2019). The GRAVY values of EtfB and EtfC proteins of E. tarda were lower indicating the proteins’ hydrophobic and nonpolar nature may favor better interaction (Gouripur et al., 2016). Valine, leucine, and isoleucine were the major essential amino acids, while alanine, serine, and glycine were common among nonessential amino acids in the target proteins of E. tarda. Table 5. Validation of 3D models of the EtfB and EtfC proteins of E. tarda using ProQ and ProSA web servers.
Fig. 2. ProSA web Z-scores, the plot of Z- scores and Ramachandran plot of the (a–c) EtfB and (d–f) EtfC proteins of E. tarda, respectively, generated by protein structure analysis. As the proteins were soluble, the transmembrane region of the proteins could not be detected. The CYS_REC tool revealed that both proteins of E. tarda have a limited number of cysteines in their amino acid sequences. The disulfide bond formed by cysteine is a measure of protein stability. Since none of the cysteines formed disulphide bonds indicating that these proteins are not strong. Most amino acids contributed to the random coils in both proteins. Though alpha helix and beta sheets play a major role in the tertiary structure of a protein. The alpha helix is a common pattern in the protein secondary structure and is a right-hand-helix in which each backbone N−H group hydrogen bonds to the backbone C=O. Other secondary structures like 310 helix, pi helix, beta bridge, bend region, ambiguous states, and other states were absent. Based on the similarity between the identified template and the sequence, the 3D model was built for the virulence proteins using the SWISS-MODEL server. The GMQE scores, a quality estimate that merges the target–template alignment properties (Waterhouse et al., 2018) were between 0 and 1 for EtfB and EtfC proteins. The estimated high GMQE scores proposed that the quality of the models was reliable. The QMEAN Z-scores, the composite estimator based on the various geometrical properties, were close to zero (Table 4), which showed a good concordance between the model structure and experimental structures of the same size (Benkert et al., 2008). Table 6. Top five herbal compounds with lower net binding free energy against the EtfB and EtfC proteins of E. tarda by molecular docking.
The RAMPAGE assessed the residue percentage in favored and allowed areas and the percentage in the outlier region. The outlier residues are the ones that do not have stable phi (φ) and psi (Ψ) angle values and are, therefore, predicted to be unstable. The expected residue percentages in favored and allowed regions were ~98.0% and ~2.0%, respectively (Lovell et al., 2003). The LG scores and MaxSub values (>0.10) of proteins predicted by the ProQ server indicated the fairly good quality of the models. The ProSA-web validates the protein model and represents it in different forms as a Z score. The Z-scores in the form of an image are determined from their lengths by the X-ray crystallography (light blue) and nuclear magnetic resonance spectroscopy (dark blue) (Pathak et al., 2014). The reported Z-scores indicated the overall model quality. The positive Z-score values reflect problematic portions of the input structure. The other two ways of representation rely on the predicted Z-scores. The exact position of Z-scores in the image is shown, predominantly of negative values, thus indicating good quality of the predicted models. The plot of residue scores represents the arrangement of amino acid residues and leads to an average overall quality of the models. The Ramachandran plot analysis indicated that the quality of the experimental structures was much higher, as revealed by the residues in the favored (green), allowed (light green), generously allowed (light blue), and disallowed (white) regions (Hasan et al., 2015).
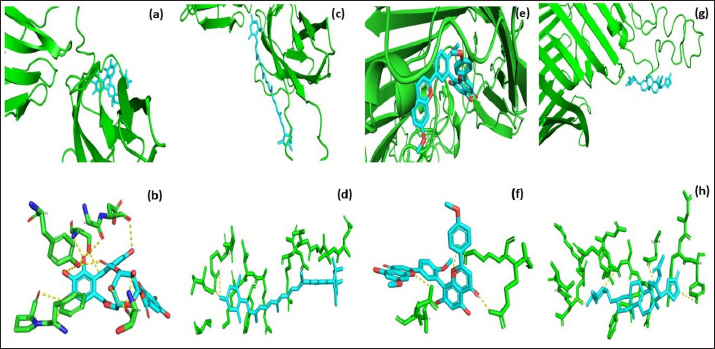
Fig. 3. Molecular docking and interaction of E. tarda virulent protein EtfB with antimicrobial herbal compounds (a) corilagin and (b) interacting amino acid residues: GLY-31, ASP-32, SER-90, PHE-94, PRO-95, TYR-131, and SER-132, (c) lutein and (d) interacting amino acid residue—ARG26, as well as virulent protein EtfC with antimicrobial herbal compounds (e) sciadopitysin and (f) interacting amino acid residues—VAL 252, ASN 254, ARG 459, and ARG 519 (g) oleandrin and (h) interacting amino acid residues—GLY 366, TYR 370, GLU 484, and TYR 488 as predicted using HDOCK server. Molecular docking has become a progressively essential tool in the discovery of drugs. A perusal of the literature indicated that corilagin plays a role as an antihypertensive, nonsteroidal anti-inflammatory, and antioxidant (Zhao et al., 2008). Lutein is an eye-protective nutrient with antioxidant properties (He et al., 2011). Isoginkgetin is involved in mRNA splicing and has been described as an anti-tumor agent (O’Brien et al., 2008). Nerium indicum (oleander) is employed in conventional medicine to treat diseases such as cardiac illnesses, asthma, cancer, and epilepsy. It has antibacterial, antiviral, antioxidant, neuroprotective, cardiotonic, and antidiabetic properties (Dey and Chaudhuri, 2014). The docking results indicated that these antibacterial herbal compounds from ink nut (T. chebula) and nutgrass (C. rotundus) may control edwardsiellosis in aquaculture. The molecular docking and interactions between the virulence proteins of E. tarda (EtfB and EtfC) and ligands such as corilagin, lutein, sciadopitysin, and oleandrin are represented pictorially using the HDOCK server in Figure 3a, c, e, and g. The amino acid labeling of interactions between the respective ligands on the target virulence protein as predicted by the HDOCK server is depicted in Figure 3b, d, f, and h. This study, thus, provided insight into the natural alternatives to antibiotics, which is an exhilarating and promising area of research. The in-silico analysis may reduce the number of molecules to be tested through experiments, accelerate drug discovery using structural pharmaceutical properties, and ultimately save time and expenses. ConclusionThe present study investigated the characteristics of fimbrial virulence proteins of E. tarda and constructed 3D models to analyze the protein-ligand binding of virulence factors such as EtfB and EtfC against different anti-virulent herbal compounds by molecular docking to identify the best compound that can be used to combat edwardsiellosis. The docking results showed that corilagin from T. chebula binds well with EtfB protein, whereas sciadopitysin from C. rotundus binds well with EtfC protein. The in-silico characterization, prediction of 3-D structure, and comparative docking analysis provided the baseline data on the EtfB and EtfC proteins of E. tarda that would facilitate further research on the potential drug designing, effective dose, challenge study with gene expression analysis and mode of application of the identified herbal compounds against edwardsiellosis disease. The current study’s results provided a basis for understanding the role of bioinformatics in fish health management. AcknowledgmentThe authors thank the Vice-Chancellor, West Bengal University of Animal and Fishery Sciences, Kolkata, for providing the necessary facilities to carry out the work. ReferencesBenkert, P., Tosatto, S.C. and Schomburg, D. 2008. QMEAN: a comprehensive scoring function for model quality assessment. Proteins 71(1), 261–277. Cristobal, S., Zemla, A., Fischer, D., Rychlewski, L. and Elofsson, A. 2001. ProQ: a study of quality measures for protein threading models. BMC Bioinform. 2(1), 5. Devi, D. and Jo, R 2015. Effect of antibiotics on modelled novel outer membrane proteins of Edwardsiella tarda—a fish pathogen. World J. Pharm. Pharm. Sci. 4(4), 1092–1104. Dey, P. and Chaudhuri, T.K. 2014. Pharmacological aspects of Nerium indicum Mill: a comprehensive review. Pharmacogn. Rev. 8(16), 156. Fogelson, S.B., Petty, B.D. and Reichley, S.R. 2016. Histologic and molecular characterization of Edwardsiella piscicida infection in largemouth bass (Micropterus salmoides). J. Vet. Diagn. Invest. 28(3), 338–344. Gouripur, G.C., Kaliwal, R.B. and Kaliwal, B.B. 2016. In silico characterization of beta-galactosidase using computational tools. J. Bioinform. Seq. Anal. 8(1), 1–11. Guruprasad, K., Reddy, B.B. and Pandit, M.W. 1990. Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. Des. Sel. 4(2), 155–161. Hasan, M.A., Mazumder, M.H.H., Chowdhury, A.S., Datta, A. and Khan, M.A. 2015. Molecular-docking study of malaria drug target enzyme transketolase in Plasmodium falciparum 3D7 portends the novel approach to its treatment. Sour. Code Biol. Med. 10(1), 7. He, R.R., Tsoi, B., Lan, F., Yao, N., Yao, X.S. and Kurihara, H. 2011. Antioxidant properties of lutein contribute to the protection against lipopolysaccharide-induced uveitis in mice. Chin. Med. 6(1), 38. Jin, R.P., Hu, Y.H., Sun, B.G., Zhang, X.H. and Sun, L. 2012. Edwardsiella tarda sialidase: pathogenicity involvement and vaccine potential. Fish Shellfish Immunol. 33(3), 514–521. Juibari, A.D., Ramezani, S. and Rezadoust, M.H. 2019. Bioinformatics analysis of various signal peptides for periplasmic expression of parathyroid hormone in E. coli. J. Med. Life 12(2), 184. Lakowicz, J.R. 2013. Principles of fluorescence spectroscopy. New York, NY: Springer Science & Business Media. Leung, K.Y., Siame, B.A., Tenkink, B.J., Noort, R.J. and Mok, Y.K. 2012. Edwardsiella tarda-virulence mechanisms of an emerging gastroenteritis pathogen. Microbes. Infect. 14(1), 26–34. Lovell, S.C., Davis, I.W., Arendall III, W.B. De Bakker, P.I., Word, J.M., Prisant, M.G., Richardson, J.S. and Richardson, D.C. 2003. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins 50(3), 437–450. Macindoe, G., Mavridis, L., Venkatraman, V., Devignes, M.D. and Ritchie, D.W. 2010. HexServer: an FFT-based protein docking server powered by graphics processors. Nucleic Acids. Res. 38(suppl_2), W445–W449. Mishra, S.S., Das, R., Dhiman, M., Choudhary, P., Debbarma, J., Sahoo, S.N., Barua, A., Giri, B.S., Ramesh, R., Ananda, K., Mishra, C.K. and Swain, P. 2017. Present status of fish disease management in freshwater aquaculture in India: state-of-the-art-review. J. Aquac. Fisheries 1(003), 14. Moustafa, E.M., Omar, A.A.E. and Abdo, W.S. 2016. Insight into the virulence-related genes of Edwardsiella tarda isolated from cultured freshwater fish in Egypt. Worlds Vet. J. 6(3), 101–109. Neema, M., Karunasagar, I. and Karunasagar, I. 2011. In silico identification and characterization of novel drug targets and outer membrane proteins in the fish pathogen Edwardsiella tarda. Open Access Bioinform. 3, 37–42. O’Brien, K., Matlin, A.J., Lowell, A.M. and 2008 Moore, M.J. The biflavonoid isoginkgetin is a general inhibitor of Pre-mRNA splicing. J. Biol. Chem. 283(48), 33147–33154. Park, S.B., Aoki, T. and Jung, T.S. 2012. Pathogenesis of and strategies for preventing Edwardsiella tarda infection in fish. Vet. Res. 43(1), 67. Pathak, R., Narang, P., Chandra, M., Kumar, R., Sharma, P.K. and Gautam, H.K. 2014. Homology modeling and comparative profiling of superoxide dismutase among extremophiles: Exiguobacterium as a model organism. Indian. J. Microbiol. 54(4), 450–458. Ramachandran, G.N., Ramakrishnan, C. and Sasisekharan, V. 1963. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 7, 95–99. Reichley, S.R., Ware, C., Steadman, J. Gaunt, P.S., García, J.C., LaFrentz, B.R., Thachil, A., Waldbieser, G.C., Stine, C.B., Buján, N. and Arias, C.R., 2017. Comparative phenotypic and genotypic analysis of Edwardsiella isolates from different hosts and geographic origins, with emphasis on isolates formerly classified as E. tarda, and evaluation of diagnostic methods. J. Clin. Microbiol. 55(12), 3466–3491. Sakai, T., Iida, T., Osatomi, K. and Kanai, K. 2007. Detection of type 1 fimbrial genes in fish pathogenic and non-pathogenic Edwardsiella tarda strains by PCR. Fish. Pathol. 42(2), 115–117. Shakya, S.R. 2017. Effect of herbs and herbal products feed supplements on growth in fishes: a review. Nepal. J. Biotechnol. 5(1), 58–63. Waterhouse, A., Bertoni, M., Bienert, S. Studer, G., Tauriello, G., Gumienny, R., Heer, F.T., de Beer, T.A.P., Rempfer, C., Bordoli, L. and Lepore, R., 2018. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids. Res. 46(W1), W296–W303. Watts, J.E., Schreier, H.J., Lanska, L. and Hale, M.S. 2017. The rising tide of antimicrobial resistance in aquaculture: sources, sinks and solutions. Mar. Drugs 15(6), 158. Wiederstein, M. and Sippl, M.J. 2007. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids. Res. 35(suppl_2), W407–W410. Yan, Y., Tao, H., He, J. and Huang, S.Y. 2020. The HDOCK server for integrated protein-protein docking. Nat. Protoc. 15(Suppl-25), 1829–1852. Zhao, L., Zhang, S.L., Tao, J.Y., Pang, R., Jin, F., Guo, Y.J., Dong, J.H., Ye, P., Zhao, H.Y. and Zheng, G.H. 2008. Preliminary exploration on anti-inflammatory mechanism of Corilagin (beta-1-O-galloyl-3, 6-(R)-hexahydroxydiphenoyl-D-glucose) in vitro. Int. Immunopharmacol. 8(7), 1059–1064. | ||

| How to Cite this Article |
| Pubmed Style Rajeshwar BN, Abraham TJ. In silico analysis, modelling and docking studies on the etfB and etfC virulence proteins of the fish pathogen Edwardsiella tarda. J Microbiol Infect Dis. 2023; 13(4): 182-189. doi:10.5455/JMID.2023.v13.i4.3 Web Style Rajeshwar BN, Abraham TJ. In silico analysis, modelling and docking studies on the etfB and etfC virulence proteins of the fish pathogen Edwardsiella tarda. https://www.jmidonline.org/?mno=302657417 [Access: July 12, 2025]. doi:10.5455/JMID.2023.v13.i4.3 AMA (American Medical Association) Style Rajeshwar BN, Abraham TJ. In silico analysis, modelling and docking studies on the etfB and etfC virulence proteins of the fish pathogen Edwardsiella tarda. J Microbiol Infect Dis. 2023; 13(4): 182-189. doi:10.5455/JMID.2023.v13.i4.3 Vancouver/ICMJE Style Rajeshwar BN, Abraham TJ. In silico analysis, modelling and docking studies on the etfB and etfC virulence proteins of the fish pathogen Edwardsiella tarda. J Microbiol Infect Dis. (2023), [cited July 12, 2025]; 13(4): 182-189. doi:10.5455/JMID.2023.v13.i4.3 Harvard Style Rajeshwar, B. N. & Abraham, . T. J. (2023) In silico analysis, modelling and docking studies on the etfB and etfC virulence proteins of the fish pathogen Edwardsiella tarda. J Microbiol Infect Dis, 13 (4), 182-189. doi:10.5455/JMID.2023.v13.i4.3 Turabian Style Rajeshwar, Balakrishnan Naveen, and Thangapalam Jawahar Abraham. 2023. In silico analysis, modelling and docking studies on the etfB and etfC virulence proteins of the fish pathogen Edwardsiella tarda. Journal of Microbiology and Infectious Diseases, 13 (4), 182-189. doi:10.5455/JMID.2023.v13.i4.3 Chicago Style Rajeshwar, Balakrishnan Naveen, and Thangapalam Jawahar Abraham. "In silico analysis, modelling and docking studies on the etfB and etfC virulence proteins of the fish pathogen Edwardsiella tarda." Journal of Microbiology and Infectious Diseases 13 (2023), 182-189. doi:10.5455/JMID.2023.v13.i4.3 MLA (The Modern Language Association) Style Rajeshwar, Balakrishnan Naveen, and Thangapalam Jawahar Abraham. "In silico analysis, modelling and docking studies on the etfB and etfC virulence proteins of the fish pathogen Edwardsiella tarda." Journal of Microbiology and Infectious Diseases 13.4 (2023), 182-189. Print. doi:10.5455/JMID.2023.v13.i4.3 APA (American Psychological Association) Style Rajeshwar, B. N. & Abraham, . T. J. (2023) In silico analysis, modelling and docking studies on the etfB and etfC virulence proteins of the fish pathogen Edwardsiella tarda. Journal of Microbiology and Infectious Diseases, 13 (4), 182-189. doi:10.5455/JMID.2023.v13.i4.3 |

