





| Research Article | ||
J. Microbiol. Infect. Dis., (2023), Vol. 13(2): 59–71 Original Research Tropism prediction in HIV-1 variants circulating in Cuba, 2017-2019: Implications for the pathogenesis of infection and response to MaravirocAnamary Suárez Batista1†, Lissette Pérez Santos2†, Liuber Yans Machado Zaldívar1, Melissa Méndez González2, Yanet Pintos Saavedra2, Jorge Pérez Ávila2, Héctor Manuel Díaz Torres3, Tamara Rojas Núñez1, Juliet María Enriquez Puertas1, Yoanna Baños Morales2, Enrique Noa Romero1 and Vivian Kourí Cardellá2†*1AIDS Research Laboratory, San José de las Lajas, Cuba 2Institute of Tropical Medicine “Pedro Kouri”, Virology Department, IPK, La Habana, Cuba 3Hermanos Amejeiras” Clinical-Surgical Hospital, La Havana, Cuba †These authors contributed equally to this publication. *Corresponding Author: Vivian Kourí Cardellá. Institute of Tropical Medicine “Pedro Kouri”, Virology Department, IPK, La Habana, Cuba. Email: vkouri [at] ipk.sld.cu Submitted: 09/01/2023 Accepted: 16/06/2023 Published: 30/06/2023 © 2023 Journal of Microbiology and Infectious Diseases
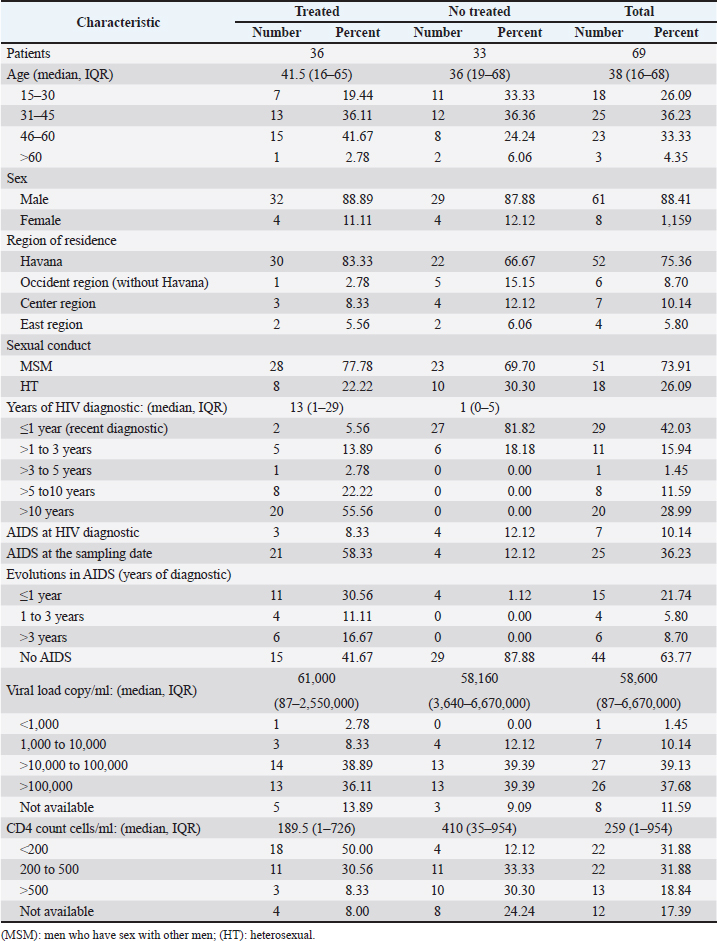
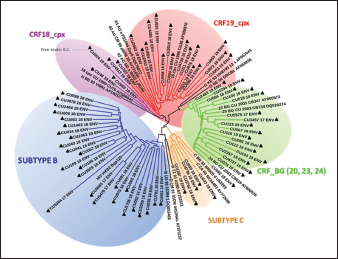
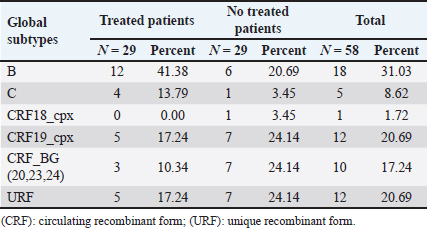
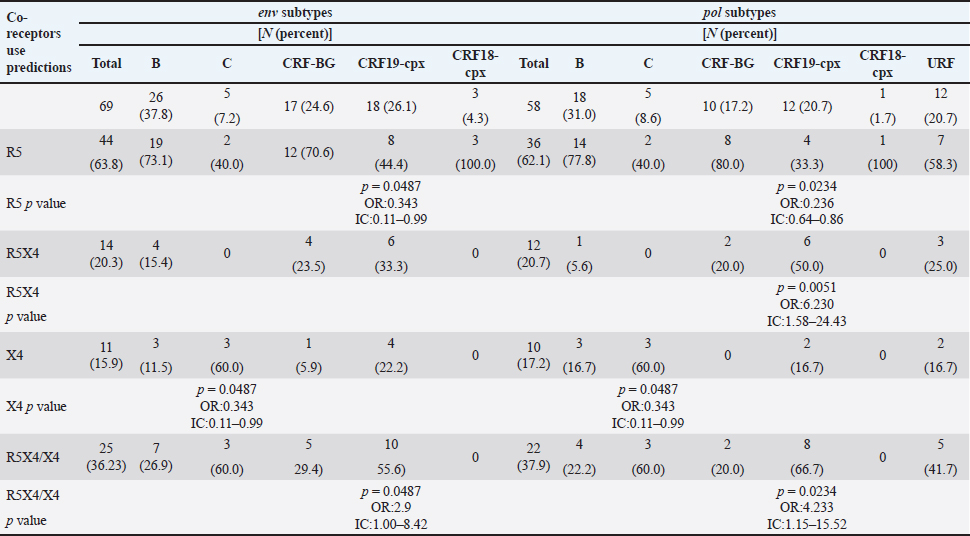
AbstractBackground: Tropism is determined by the ability of HIV-1 to bind to the receptors C-C chemokine receptor type 5 or C-X-C chemokine receptor type 4 (CXCR4) to enter the target cell, which has implications for the pathogenesis of the disease and affects treatment options. This study determined the relationship between the tropism of HIV-1 variants circulating in Cuba, with different variables, and the implications for the use of co-receptor inhibitors. Methods: Plasma samples from 69 of HIV-1 seropositive patients, period 2017–2019, were analyzed. Fragments of the env and pol regions of the virus were sequenced. The subtype and prediction of the viral phenotype, viral mutations, as well as the relationship of the phenotype and subtype with clinical, epidemiological, and virological variables were determined. Results: The recombinant CRF19_cpx (p =0.0234) showed a significant association with viruses that use the X4 and R5X4/X4 Co-receptor, even in untreated patients. Of the viruses studied, 78.26% presented at least one mutation associated with resistance to Maraviroc (MVC), although the patients had not received prior therapy with this drug. Conclusion: The preferential tropism for the CXCR4 co-receptor, detected in the CRF19_cpx variant, accompanied by greater viral replication and unrelated to the time of diagnosis of the patients, reinforces the hypothesis that the viral variant is more pathogenic. The high frequency of polymorphisms and mutations that confer resistance to MVC in the V2 and V3 regions of HIV-1 of the Cuban recombinant forms indicate that there could be a natural resistance to this antiretroviral. Keywords: HIV-1, Maraviroc, Tropism, CXCR4, CRF19_cpx. IntroductionThe variability of the HIV env gene, which plays an important role in infection and antigen recognition, has been the main obstacle to the development of vaccine candidates and new therapeutic strategies. The V3 loop, present in the env gene, is a component of the chemokine receptor-binding region and is the major determinant of Co-receptor usage. The functional importance of V3 lies in the fact that a deletion in it can completely alter the infectivity of the virus. In contrast, V2 is not essential for infectivity; however, it participates in various functions of the env gene associated with escape from neutralization mediated by V3-specific antibodies, due to changes in its length and glycosylation patterns (Zolla-Pazner and Cardozo, 2010). Viral variants that use the C-C chemokine receptor type 5 (CCR5) are characterized in cell cultures for being non-inducing syncytia, having a low replication rate, tropism for macrophages, and predominating in the early stages of infection, being classified as viruses with the R5 phenotype. Those viruses that use the C-X-C chemokine receptor type 4 (CXCR4) for cell entry, mainly infect T cells and stand out for their ability to induce syncytia, have a high replication rate, and predominate in the late stage of the disease, classifying as virus with phenotype X4. At the same time, there is a group of viruses that are classified as having a dual phenotype (R5X4) due to their ability to bind to both the CCR5 and CXCR4 co-receptors. This is considered to be a transitory phase in the evolution of the R5 virus to X4, although phenotypically they behave like X4 viruses (Sede et al., 2014). HIV-1 has demonstrated its ability to change tropism during the course of the disease, a process that the virus adaptively uses to spread in the presence of antivirals (Smyth et al., 2012). Limited access to antiretrovirals (ARVs) in some parts of the world, suboptimal levels of adherence, resistance, and failure of therapy are some of the existing problems that need to be addressed in the management of HIV infection (Aves et al., 2018). Maraviroc (MVC) is the only drug from the group of co-receptor antagonists approved by the Food and Drug Administration of the United States for the treatment of infection with R5 variants of HIV-1. It is the first ARV to target a host protein rather than a viral protein. Its mechanism of action consists of allosteric inhibition of the union of the glycoprotein gp 120 to the co-receptor CCR5, which prevents the virus from entering the cell (Gulick et al., 2008). Although this drug began to be used in 2007, the existence of genetic variants of HIV-1 resistant to MVC has already been reported. Although the mechanisms by which this occurs are not yet clear, it is known that a group of mutations in the V3 loop of the glycoprotein gp 120 are associated with resistance (Westby, 2007). In Cuba, the first HIV cases were diagnosed in 1986. Data from the National Direction of Epidemiology of the Ministry of Public Health (MINSAP, 2020) show that as of December 2021, 37,028 people were infected with HIV. The male sex is the most affected and accounts for the majority of cases 80.8%. Men who have sex with other men (MSM) represent 88.2% of the male sex, and 92.9% of people living with HIV are under treatment (MINSAP, 2020). It is known that the HIV-1 epidemic in Cuba is atypical compared to the rest of America and the Caribbean, where subtype B predominates. A high diversity of HIV-1 genetic variants, including a high rate of circulating recombinant forms (CRFs) characterizes the epidemic in Cuba. Over the last decade, the detection of the viral variation CRF19 cpx has been associated with rapid progression to AIDS, high viral load values, the X4 viral phenotype, and higher levels of RANTES in patients with less than 3 years since diagnosis (Kouri et al., 2015). The objective of this study was to determine the relationship between the tropism of HIV-1 variants circulating in Cuba, in treated and naive patients, with epidemiological, viral, and clinical variables and the implications for the use of co-receptor inhibitors. Materials and MethodsStudy universeThe universe consisted of 69 plasma samples from HIV-1 seropositive patients, 36 untreated and 33 treated with ARVs, received at the Civil Defense Scientific Research Center or at Pedro Kourí Institute for antiretroviral resistance studies between 2017 and 2019. Amplification and partial sequencing of the pol and env genesHIV ribonucleic acid (RNA) was extracted from 1 ml of plasma using the commercial QIAamp Viral RNA minikit (QIAGEN, Heidelberg, Germany). The pol gene was amplified and sequenced using the protocol described by Aleman et al. (2015). It encompasses a fragment of 1,302 bp, 297 nucleotides (99 amino acids) of protease (PR), and 1,005 nucleotides (335 amino acids) of HIV-1 reverse transcriptase (Aleman et al., 2015). The amplification and partial sequencing of the env gene (675 bp that included the V2C2V3C3 regions, from aa 158 to 385 of gp 120) were performed following the protocol described by Van Laethem et al. (2005). Sequence editing, identification of viral subtypes, and phylogenetic analysisThe sequences obtained from each of the sequenced HIV fragments were assembled and edited using the SequencherTMVersion 4.10 program (Genes Codes Corporation, USA), and the nucleotide sequence of the HIV-1 strain B.FR.83.HXB2_LAI_IIIB_BRU was used as reference. In the end, consensus sequences of the sequenced regions (pol gene, env gene) of HIV-1 were obtained from each patient analyzed. HIV subtyping tools: COMET version 2 (available at (https://comet.lih.lu) and REGA subtyping tool version 3 (available at https://rega.kuleuven.be/cev/avd /software) were used to perform the initial subtype classification from the edited sequences for each region of the virus. Since the Cuban epidemic is characterized by a high number of CRFs and unique recombinant forms (URFs), all assignments were confirmed with a phylogenetic analysis of the sequences obtained for each analyzed gene. The sequences of the pol and env genes were examined using the automated bioinformatics tool NGPhylogeny (https://ngphylogeny.fr) (Lemoine et al., 2019). Viral tropism was predicted using the G2P genotyping tool version 2.5 [co-receptor] (http://coreceptor.geno2pheno.org/). Based on the German guidelines, tropisms were divided into three groups according to the probability of incorrectly identifying an R5 virus as X4 (Vandekerckhove et al., 2011). Mutations that confer a change in viral tropism were analyzed manually, based on the net load and the 11/25 rule. MVC resistance-associated mutations in the V3 loop were manually identified, as published in the literature (Sierra et al., 2007). The analysis was also done for the V2 loop. In addition, the N glycosylation sites for both regions of gp 120 (V2 and V3) and their possible relationship with tropism and viral subtype were identified (Utachee et al., 2010). Statistical analysisA database was created in Excel. Mean, SD, median, interquartile range, and frequencies (%) were used to describe patient characteristics. X2 and Fisher’s tests were used to compare categorical and continuous variables. The odd ratios (OR) and their 95% confidence interval (CI) were estimated. A value of p < 0.05 was considered statistically significant. All statistical analyzes were performed using SPSS statistical software version 18 (SPSS Inc., Chicago, IL). To make comparisons between quantitative variables, the Anova tests with Dunnett and Mann Whitney post-test (Graphpad Prism 5 Program, San Diego, CA) were used. Ethical approvalThe study was conducted following the latest revision of the Declaration of Helsinki and the ethical, institutional, and regional standards of current medicine. The individuals, after receiving all the necessary information through talks and written material, decided whether they would participate in the study or not, by signing and informed consent. ResultsGeneral characteristics of the patients studiedOf the 69 samples of HIV-1 seropositive patients studied, corresponding to the period 2017–2019, 33 belonged to treated patients and 36 to untreated patients; therefore, this condition was related to the rest of the variables analyzed (Table 1). Male patients account for 88.41%, of which 73.91% were MSM. Significant differences were observed between treated and untreated patients in terms of years of diagnosis and progression to AIDS. Of the treated patients, 55.56% had been diagnosed more than 10 years ago, while in the untreated group, 100% of the patients were in the range of ≤1–3 years, and 81.82% had newly diagnosed HIV infection. The overall AIDS frequency was 36.23%, of which only 10.14% were already AIDS cases at the time of diagnosis, being 10 times more frequent than treated patients had AIDS at the time of sample collection compared with the untreated patients (58.33% vs. 12.12%, p =0.0001, OR:10.15, CI:2.94–34.99). The median viral load of HIV in the patients studied was high (58,600 copies/ml) and no differences were found between the treated and untreated groups, with the highest percentages being observed in the ranges of 10,000–100,000 copies/ml. and greater than 100,000 copies/ml. TCD4 + cell count was different between treated and untreated patients. Fifty percent of treated patients had <200 cells/ml compared to 12.12% of untreated patients (p=0.0007, OR: 7.25, CI: 2.11–24.87). On the other hand, 30.30% of untreated patients exhibited CD4 count greater than 500 cells/ml, compared to 8.33% of treated patients (p=0.0206, OR: 4.78, CI: 1.18–19.31). Detection of HIV-1 subtypes in the pol and env regionsIn the present study, 69 samples were used for the phylogenetic analysis, which was conducted in a region of 187 aa of the env gene that encodes the C2V3C3 fragments of gp 120. Additionally, it was possible to sequence and analyze a region of the pol gene (99 aa from PR and 335 from TI) in 58 of the 69 samples (84.05%). When studying the env and pol genes separately and analyzing the viral variants globally, taking into account the joint analysis of the subtypes detected in the two regions of the virus, the most often detected HIV-1 subtypes were subtype B, CRF19_cpx, and CRF_BG (20, 23, 24) (Figs. 1 and 2; Table 2). HIV-1 subtypes did not show significant differences between treated and untreated patients, although subtypes B and C were more frequently detected among treated patients; and CRF_BG, CRF19_cpx, and URF among untreated patients (Table 3). When analyzing the subtypes globally, differences were found in 12 patients, who presented a different viral subtype between the pol and env genes. These viral variants were considered as URFs (five in treated patients and seven in untreated patients), representing 20.7% of the viral variants detected when analyzing the subtype globally. It is noteworthy that, in the present study, the CRF18_cpx virus was more represented as part of the URF (7/12, 58.3%, p=0.0000, OR: 63, CI: 6.3–622) and specifically among untreated patients, in contrast to treated patients (5/7 vs. 2/5, p =0.0455, OR: 21.00, CI: 1.21–564.14). The most frequent URF was the combination of CRF18_cpx/CRF19_cpx, observed among untreated patients (42.8%). Prediction of viral co-receptor usage and relationship to HIV-1 subtypeWhen investigating the prediction of the use of the co-receptor in this study, it was observed that the R5 phenotype was present in the majority of the samples, when analyzing the subtype both in the env fragment (63.77%, 44/69 samples), and in the global subtype (62.06%, 36/58 samples), while 36.23% and 37.93% were R5X4/X4 for the same fragments, respectively (Table 4). However, it was found that the viral variants corresponding to subtypes C and CRF19_cpx showed a significant association with the use of the co-receptor CCR5 and CCR5/CCRX4, respectively. When stratifying co-receptor usage between treated and untreated patients, no differences were observed between the two groups. However, when analyzing the env gene related to the use of the co-receptor, some differences between the subtypes were revealed. Subtype C viruses were predominantly X4 in treated patients (3/4, 75%, p=0.0178, OR: 14.777, CI: 1.71–127.58) and CRF19_cpx type R5X4 viruses (3/7, 42.86%, p=0.0375, OR: 5.88, CI: 1.09–32.74). In untreated patients, it is striking that although most of these patients (81.82%) had been recently diagnosed, those infected with the CRF19_cpx virus were mostly type X4 (4/7 patients, 36.36%, p=0.0081, OR: 27.00, CI: 1.29–562.39). Furthermore, three of these patients had been diagnosed with HIV infection less than a year ago and the fourth had been diagnosed 2 years ago, which would support the previously described hypothesis that the X4 tropism observed for the CRF19_cpx virus is not related to infection evolution of the natural history of HIV, but to the rapid progression to AIDS. However, as we are not certain of the date of infection of these patients, it cannot be ruled out that they are late diagnoses of HIV infection. Table 1. Epidemiological, clinical, and immunological characteristics of the patients studied.
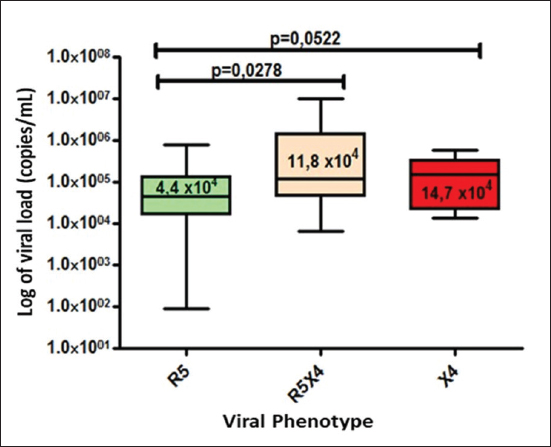
Fig. 1. Phylogenetic tree of the HIV-1 pol gene. The analysis included 87 nucleotide sequences, 58 sequenced in the present study and 29 referenced. The evolutionary history was inferred using the maximum likelihood method (PhyML version 3) and the GTR nucleotide substitution model. The inferred consensus tree was used to represent the evolutionary history of the analyzed sequences. The different subtypes are shown in a circular tree with branches indicated in different colors according to the subtype: Pink, subtype A; Blue, subtype B; Orange, subtype C; Grey, subtype G; Green, CRFs_BG (20, 23,24), Red, CRF19_cpx; Violet, CRF18_cpx. The Cuban samples sequenced in this study are marked with a black triangle, the rest are reference sequences of the different subtypes of HIV-1, included in this work. In contrast, subtype C (also associated in this study with the X4 phenotype) was found mainly in treated patients, most of whom had been diagnosed 5 years or more ago, which coincides with what has been classically established for the change in tropism from R5 to X4. Relationship of the viral phenotype with clinical and epidemiological variables, viral load, and TCD4+ cell countSeveral factors can determine the progression of AIDS in patients: the host, the virus, and the environment. In the present study, no significant differences were observed between the epidemiological variables and the viral phenotype. Of the total number of patients who had viral load available (61/69), 24 were classified as R5X4/X4. Patients infected with these viral phenotypes are 3.9 times more likely to have a viral load greater than 100,000 copies/ml (p=0.012, OR: 3.93, CI: 1.32–11.67). In addition, when examining the median viral loads (Fig. 3) in relation to the viral phenotype, higher viral load values were observed for the R5X4 and X4 phenotypes, this difference is significant for the R5X4 viruses ( p=0.0278). However, when treated and untreated patients were analyzed separately, no significant differences were observed (data not shown). Although the median CD4 counts were higher in patients infected with the R5 virus, the difference was not significant when studying the total number of samples with available CD4.
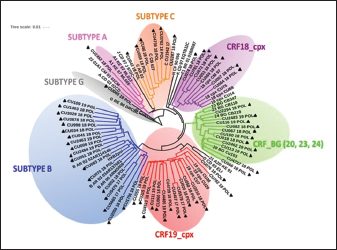
Fig. 2. Phylogenetic tree of the HIV-1 env gene. The analysis included 86 nucleotide sequences, 69 sequenced in the present study and 17 referenced. The evolutionary history was inferred using the maximum likelihood method (PhyML version 3) and the GTR nucleotide substitution model. The inferred consensus tree was used to represent the evolutionary history of the analyzed sequences. The different subtypes are shown in a circular tree with branches indicated in different colors according to the subtype: Blue, subtype B; Orange, subtype C; Green, CRFs_BG (20, 23,24), Red, CRF19_cpx; Violet, CRF18_cpx. The Cuban samples sequenced in this study are marked with a black triangle, the rest are reference sequences of the different subtypes of HIV-1, included in this work. Table 2. Variants of HIV-1 obtained by partial sequencing of two regions of the virus, in the analyzed samples.
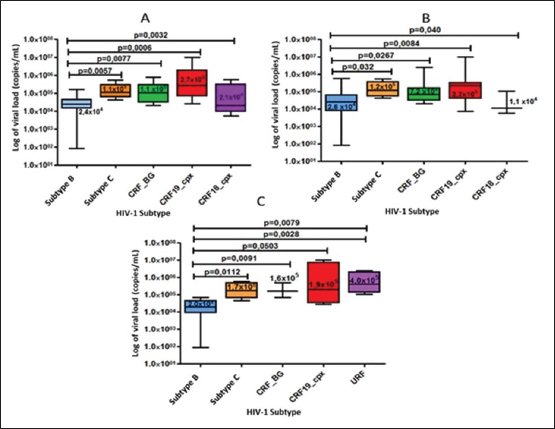
When comparing the viral load values between the different viral subtypes, taking into account the variant detected when sequencing the env gene (Fig. 4A), pol (Fig. 4B) and in the analysis of the global subtype (Fig. 4C), differences were observed in the viral load values between the subtypes, demonstrating significantly higher viral load figures for the CRF19_cpx viruses, compared to the other viruses (p ≤ 0.05). On the other hand, subtype B showed significantly lower values of viral load, in relation to the other subtypes. Table 3. Global HIV-1 subtype in treated and untreated patients by partial sequencing of two regions of the virus.
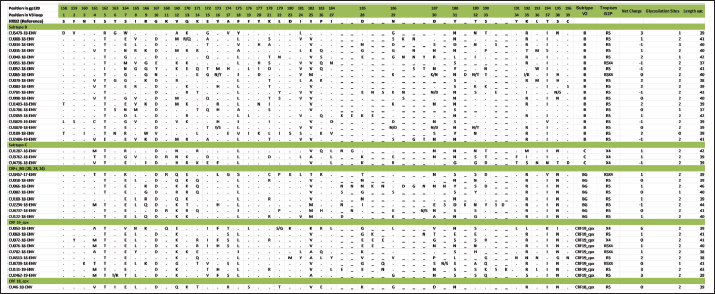
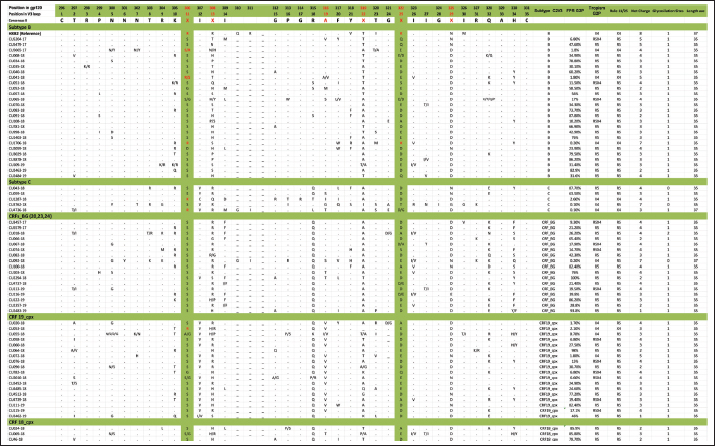
Mutations in the V2 and V3 regions of the envelope protein gp 120, in the Cuban sequence s of HIV-1 mutations in the V2 and V3 loopA comparison of the amino acid sequences of the V3 loop among the 69 samples showed significant differences between the different subtypes. In 40 (57.97%) of the 69 sequences of the env gene, it was possible to analyze the V3 region and V2 variable region. The length of the sequences varied between the samples studied, being CU051-18 the shortest (37 amino acids, subtype B, R5X4 tropism), and CU066-18 the longest (46 amino acids, CRF_BG, R5 tropism). The average length of the sequences was 40.05 amino acids with multiple deletions and insertions, particularly in the hypervariable region. Similar to what was observed for V3, some differences related to the viral subtype were observed in the amino acid sequence (Figs. 5 and 6). Mutations in V2 and V3 are associated with a change in tropism and resistance to MVCWhen analyzing the V3 loop sequences of the 69 samples studied, we were able to observe that, in general, 54 (78.26%) presented at least one mutation associated with resistance to MVC. In the present investigation, the X13H mutation, also called P/T308H, (depending on whether only the position from the beginning of the V3 loop or gp 120 is taken into account) was frequently detected in almost all the viral variants studied, except in subtype C, where I323V was found, although to a lesser extent. The A316T mutation was also present in the non-B subtype samples (except for CRF19_cpx). The G11R mutation was detected in subtype B and C samples (p=0.012 OR: 10.36 CI: 1.15–92.90), while the X13R mutation was frequently observed in non-B subtype samples (p=0.0004 OR: 32.47 CI: 1.89–556.37). The change from A to K at position 25 (A25K) was only observed in a few samples, all of which were subtype B (Figs. 5 and 6). It was shown that the X13R mutation was more frequent in untreated patients than in treated patients, 17/33 (51.52%) (p=0.044, OR: 2.76, CI: 1.02–7.50), and for the rest of the described mutations no significant differences were found. In the V2 region, an extensive bibliographic search was carried out to investigate the mutations related to resistance to co-receptor inhibitors that have been described in various studies. We identified the V169M mutation, detected in five samples of the present study (CU008-18, CU035-18, CU998-18, CU1403-18, and CU2484-19), all of subtype B and with CCR5 tropism. The N192K mutation was also detected in another five samples (CU052-18, CU065-18, CU083-18, CU1706-18, and CU3029-18), all also of subtype B and with variable tropism: three R5, one R5X4, and one X4. Glycosylation patterns in the V3 and V2 loopsThe average of the glycosylation sites found in the V2 and V3 loops of the sequenced Cuban samples was 1.85 and 0.97, respectively. In 66 of 69 V3 loop sequences (95.65%), a single glycosylation site was identified (positions 6–8 of the V3 loop). Only two subtype C samples (CU043-18, CU3762-18) and one CRF_BG (CU092-18) did not show this pattern. Sample CU018-18 presented an additional glycosylation site at positions 32–34 of the V3 loop. Of the 40 samples that underwent V2 analysis, 82.5% (33/40) had two glycosylation sites, 2.5% (1/40) had none or three sites, and 12.5% (5/40) presented only one. Except for four samples (CU5479-17, CU109-19, CU2484-19, and CU4739-18), the rest had a glycosylation site at positions 3–5, but the majority of the glycosylation sites were found at positions 185–190, being mostly the NNXS(T) type pattern. Additionally, two of the samples had patterns at positions 34–38 of V2. When analyzing the net charge, values lower than those reported for the V3 loop (average 1.22) were observed, probably because there is a greater number of negatively charged amino acids (D and E). In our study, no significant differences were detected in the glycosylation patterns related to tropism, or with the viral subtypes. Table 4. Comparison of prediction of co-receptor use between detected HIV subtypes.
Fig. 3. Median viral load values stratified according to co-receptor use.
Fig. 4. Comparison of viral load values according to the viral variant detected in the pol gene region (A), in the env gene (B), and in the global subtype (C) of the samples studied.
Fig. 5. Alignment of the amino acid sequence of the V3 loop of the HIV-1 gp 120 protein and prediction of viral tropism.
Fig. 6. Alignment of the amino acid sequence of the V2 loop of the HIV-1 gp 120 protein. DiscussionIn Cuba, despite the low prevalence of HIV-1 infection, a great variety of subtypes circulate (Kouri et al., 2012), mainly B, C, G, and H, the recombinant forms CRFs_BG (20, 23, and 24) and the complex recombinants CRF19_cpx and CRF18_cpx (Perez et al., 2006). While the BG recombinants are of Cuban origin, the CRF18_cpx and CRF19_cpx appear to be of African origin (Perez et al., 2006), although they behave epidemically only in Cuba. This diversity contrasts with the almost exclusive epidemic of subtype B in the rest of the Caribbean (94%) (Kouri et al., 2012). In a study carried out by Kourí et al. (2019) where more than 1,500 sequences of HIV-seropositive patients were analyzed, with dates of diagnosis of the infection from 1986 to 2018, it was observed that subtype B was found in 30% of the population studied, while the recombinant forms (CRF_BG, CRF19_cpx, and CRF18_cpx) were found in 51% of the samples. To a lesser extent, other subtypes were found, such as A, C, F, G, H, J, other CRFs, and URFs. HIV-1 subtypes did not change significantly between treated and untreated patients, although subtypes B and C were more frequently detected among treated patients; and CRF_BG, CRF19_cpx, and URF among untreated patients. Treated patients had been diagnosed with HIV long before untreated patients, which explains the differences observed in the frequencies of the subtypes. In the last two decades, a decrease in the circulation of subtype B has been observed at the expense of an increase of recombinant BG viruses (CRFBG_20, CRFBG_23, and CRFBG_24) and CRF19_cpx (Najera et al., 2002; Perez et al., 2006; Kouri et al., 2012, 2019). Najera et al. (2002) in an article published in the 2000s, discuss the under-registration of URFs in areas where multiple subtypes and CRFs circulate at the same time, as well as the possibility of generating new genetic forms in those populations where this occurs. This phenomenon coincides with that observed in the Cuban HIV epidemic, where a large number of genetic variants circulate, so an increase in URF in Cuba is to be expected (Machado et al., 2017). This result also warns about the importance of sequencing more than one HIV gene to obtain more precise results when conducting molecular epidemiology studies in the country. It would be worth investigating in the future whether the frequent finding of CRF18_cpx as part of the URF could be related to an evolutionary disadvantage of this viral variant and that, in combination with another subtype or another recombinant form; it could increase its replicative capacity and its pathogenicity. In a previous study, where the use of the co-receptor was studied in 42 Cuban samples, it was observed that the two samples identified as CRF18_cpx had CCR5 tropism (Kourí et al., 2016). A change in viral tropism is known to be associated with accelerated deterioration of the immune system and poor clinical prognosis. Furthermore, it has been observed that treatment with CCR5 antagonists can select minority populations of viruses with the ability to utilize CXCR4. That is why the determination of tropism is important, not only from the clinical point of view, for the patient who will start therapy with MVC, but also from the epidemiological point of view, to identify the viruses that potentially have greater virulence and that could be related to an increased risk of rapid progression to AIDS (Dean, 2012). The literature reports that 60% to 80% of subjects, in the early stages of HIV infection, carry R5 viral variants and they are considered to be the main viral variants responsible for new infections. Viral variants X4 or R5X4 emerge in approximately 50% of individuals in the later stages of the disease, generally associated with rapid depletion of TCD4+ cells and progression to AIDS (Oberle et al., 2016). On the other hand, Kourí et al. (2016) published a study of 42 samples from HIV-1 seropositive Cuban patients and they described that 38.1% of the patients carried viruses with reduced capacity for the use of co-receptor CCR5 (R5X4 or X4 virus), which coincides with the findings of the present investigation. The first report on the relationship of CRF19_cpx with the use of co-receptor CXCR4 was carried out in samples of untreated Cuban patients collected between 2007 and 2011 (Kouri et al., 2015). That study found that CRF19_cpx was associated with rapid progression to AIDS, and one of the hypotheses to explain this rapid progression was the finding of higher levels of RANTES expression, higher viral load, and a significantly more frequent proportion of X4 viruses among patients infected with this viral variant (Kouri et al., 2015). Another later study also showed that CRF19_cpx viruses presented strains with R5X4/X4 (p=0.009) or X4 (p=0.019) tropism more frequently (Kourí et al., 2016). The results of the present investigation agree with the above and reinforce the theory that the CRF19_cpx virus could have a high replicative capacity and greater pathogenicity. The association of subtype C viruses with viruses that use co-receptor CXCR4, obtained in the present investigation, contrasts with previous studies. In ex vivo studies, some authors have published that, although subtype C is the most prevalent worldwide, paradoxically it seems less virulent than other subtypes (Abraha et al., 2009), suggesting a single molecular mechanism that simultaneously affects the replicative efficiency of the virus while facilitating viral transmission events. Many mechanisms seem to involve Env glycoproteins and entry mechanisms, observing that the detection of R5X4 and X4 variants is a relatively rare event for subtype C, even in advanced stages of infection, when compared to subtype B (Jakobsen et al., 2013). The most common mutations that have been reported in the V3 loop are: substitutions of glycine (G) for (R) at position 11 (G11R), proline (P) for R at position 13 (P13R), and alanine (A) for K at position 25 (A25K) (Barmania and Pepper, 2013). Other in vitro studies have shown that mutations in this region do not usually confer complete resistance to MVC, but instead require the cooperation of several mutations (Asin-Milan et al., 2013). The specific polymorphisms of the different subtypes coincide with the recently published findings in Cuban sequences, where a pattern of mutations was observed for each of the different viruses that circulate in Cuba (Kourí et al., 2016). Other researchers have also previously reported the association between some viral variants and certain V3 loop mutations. Zhang et al. (2014) found that the E25R/Q/N mutation was the most frequent among subtypes C, B, and CRF07_BC. V3 sequences have been reported to vary considerably between different viral strains and subtypes, and this variability is most pronounced at the crown of the V3 loop (14 amino acids in the middle of V3). However, there are conserved regions related to the important function of V3 as an element of the chemokine receptor site (CCR5 and CXCR4) that facilitate the entry of the virus into the cell (Zolla-Pazner and Cardozo, 2010). These conserved regions are considered potential targets for the design of immunogens that are capable of generating a broad and crossed response of neutralizing antibodies against different viral strains. That is why the recognition of the regions that are specific to each viral variant and the conserved regions of the V3 loop is of great importance, specifically for the design of Cuban vaccine candidates, where you can find a great variability of subtypes, CRF and URF (Perez et al., 2006; Kouri et al., 2012, 2019). There are no previous studies from Cuba where V2 sequences have been compared between different viral variants, so this would be the first analysis detailing the V2 sequence in Cuban samples. Similar to what was observed for V3, subtype-specific amino acid patterns were detected; however, in V2 a greater variation between amino acid sequences and their length was also observed. The high variability observed in the V2 loop of the present study, both in amino acid patterns and in the presence of multiple deletions and insertions, coincides with what is reported in the literature, which states that the V2 loop is a more variable region than V3, probably because this variability does not affect viral infectivity (Zolla-Pazner and Cardozo, 2010). Two main mechanisms have been described that contribute to the emergence of resistance to MVC: the selection of minor X4 or R5X4 strains that are not inhibited by Co-receptor antagonists and the acquisition of the ability of R5 viruses to interact with the MVC-bound CCR5 Co-receptor conformation. This latter scenario typifies genuine MVC resistance (Flynn et al., 2017). Although resistance to MVC is associated with a wide variety of amino acid changes in both gp120 and gp41, changes in the V3 loop have been identified as major contributors to the resistance phenotype to mutations associated with resistance to MVC related to the viral subtype have been described by other authors. It has been suggested that natural mutations to MVC might be more common in subtype C than in subtype B of HIV-1. Analysis of 65 samples showed that 52.3% (75% of subtype C and 18.2% of subtype B) had at least one mutation associated with resistance to MVC. In 67.8% of subtype C samples and 18.2% of subtype B samples, the A316T mutation was commonly seen in the gp 120 region. A high prevalence of the A316T mutation was also found in Zambian mother-to-child couples infants infected with HIV-1 subtype C (Borm et al., 2016; Palmer and Poon, 2019). This coincides with the findings of the present study, where the A316T mutation was observed in patients infected with CRF18_cpx, CRF_BG, and subtype C, while any patient infected with subtype B presented it. The present study has the limitation that the sequencing process carried out was with the Sanger method, which detects the majority of variants present in a proportion greater than 20%. Perhaps, if we used deep sequencing, we could detect other minor variants with non-R5 tropism. In a study carried out by Palmer and Poon (2019) in which 6,605 sequences from the Los Álamos database were included, they observed that the average number of glycosylation sites for the V3 loop was 0.9, which coincides with that obtained in the present investigation, not being so in the case of the V2 loop, which was 1.85, compared to the 2.1 obtained by the aforementioned authors. Palmer and Poon (2019) determined that subtypes B and C had a significantly higher number of glycosylation sites within the V3 loop, 0.96 (0.87–1.05) and 0.95 (0.96–1.05) respectively, which does not coincide with what was observed in this study. Subtype C showed lower values than previously reported and the rest of the subtypes studied showed the same number of glycosylation sites. A study that analyzed the gp120 of the recombinant CRF01_AE of several strains that showed resistance or susceptibility to viral neutralization against an anti-CD4 monoclonal antibody, revealed that those strains that had a particular glycosylation residue in amino acids 186 and 197 (V2 region) showed resistance to neutralization (Utachee et al., 2010). As previously mentioned, many of the samples of the current investigation showed glycosylation sites at position 186 (four B subtypes, one CRF_BG, and five CRF19_cpx). It would be necessary to explore in future research whether the resistance or susceptibility of some of these strains to neutralization would have important implications for the design of vaccines or therapies. The results for position 197 are not shown, since the sequences included in this work only reached position 196. In our study, no significant differences were detected in the glycosylation patterns related to tropism or viral subtypes. The genetic diversity of HIV-1, observed in the present study, confirms the findings on the molecular epidemiology of this virus in Cuba. The preferential tropism for the co-receptor CXCR4, detected in the CRF19_cpx variant, accompanied by greater viral replication and unrelated to the time of diagnosis of the patients, reinforces the hypothesis that this viral variant is more pathogenic. The frequent finding of CRF18_cpx as part of the URF, with R5 tropism and low viral load suggests the possible intersubtype recombination with a virus with greater replicative capacity as a mechanism to increase its pathogenicity and transmissibility. The high frequency of polymorphisms and mutations that confer resistance to MVC in the V2 and V3 regions of HIV-1 of the Cuban recombinant forms indicate that there could be a natural resistance to this antiretroviral. Conflict of interestThe authors declare that there is no conflict of interest. ReferencesAbraha, A., Nankya, I.L., Gibson, R., Demers, K., Tebit, D.M., Johnston, E., Katzenstein, D., Siddiqui, A., Herrera, C., Fischetti, L., Shattock, R.J. and Arts, E.J. 2009. CCR5- and CXCR4-tropic subtype C human immunodeficiency virus type 1 isolates have a lower level of pathogenic fitness than other dominant group M subtypes: implications for the epidemic. J. Virol. 83(11), 5592–5605. Aleman, Y., Vinken, L., Kouri, V., Perez, L., Alvarez, A., Abrahantes, Y., Fonseca, C., Pérez, J., Correa, C., Soto, Y., Schrooten, Y., Vandamme, A.M. and Van Laethem, K. 2015. Performance of an in-house human immunodeficiency virus type 1 genotyping system for assessment of drug resistance in Cuba. PloS One 10(2), e0117176. Asin-Milan, O., Chamberland, A., Wei, Y., Haidara, A., Sylla, M. and Tremblay, C.L. 2013. Mutations in variable domains of the HIV-1 envelope gene can have a significant impact on maraviroc and vicriviroc resistance. AIDS. Res. Ther. 10, 15. Aves, T., Tambe, J., Siemieniuk, R.A.C. and Mbuagbaw, L. 2018. Antiretroviral resistance testing in HIV-positive people. Cochrane. Database. Syst. Rev. 11(11), CD006495. Barmania, F. and Pepper, M.S. 2013. C-C chemokine receptor type five (CCR5): an emerging target for the control of HIV infection. Appl. Transl. Genom. 2, 3–16. Borm, K., Jakobsen, M.R., Cashin, K., Flynn, J.K., Ellenberg, P., Ostergaard, L., Lee, B., Churchill, M.J., Roche, M. and Gorry, P.R. 2016. Frequency and Env determinants of HIV-1 subtype C strains from antiretroviral therapy-Naïve subjects that display incomplete inhibition by maraviroc. Retrovirology 13(1), 74. Dean, L. 2012. Maraviroc therapy and CCR5 genotype. In Medical genetics summaries. Eds., Pratt, V., McLeod, H., Rubinstein, W., Dean, L., Kattman, B. and Malheiro, A. Bethesda, MD: National Center for Biotechnology Information. MINSAP. 2020. Base de datos nacional de la infección por VIH. Dirección Nacional de Epidemiología. Flynn, J.K., Ellenberg, P., Duncan, R., Ellett, A., Zhou, J., Sterjovski, J., Cashin, K., Borm, K., Gray, L.R., Lewis, M., Jubb, B., Westby, M., Lee, B., Lewin, S.R., Churchill, M., Roche, M. and Gorry, P.R. Analysis of clinical HIV-1 strains with resistance to maraviroc reveals strain-specific resistance mutations, variable degrees of resistance, and minimal cross-resistance to other CCR5 antagonists. AIDS. Res. Hum. Retroviruses. 33(12), 1220–1235. Gulick, R.M., Lalezari, J., Goodrich, J., Clumeck, N., DeJesus, E., Horban, A., Nadler, J., Clotet, B., Karlsson, A., Wohlfeiler, M., Montana, J.B., McHale, M., Sullivan, J., Ridgway, C., Felstead, S., Dunne, M.W., van der Ryst, E. and Mayer H. 2008. Maraviroc for previously treated patients with R5 HIV-1 infection. N. Engl. J. Med. 359(14), 1429–1441. Jakobsen, M.R., Cashin, K., Roche, M., Sterjovski, J., Ellett, A., Borm, K., Flynn, J., Erikstrup, C., Gouillou, M., Gray, L.R., Saksena, N.K., Wang, B., Purcell, D.F.J., Kallestrup, P., Zinyama-Gutsire, R., Gomo, E., Ullum, H., Østergaard, L., Lee, B., Ramsland, P.A., Churchill, M.J. and Gorry, P.R. 2013. Longitudinal analysis of CCR5 and CXCR4 usage in a cohort of antiretroviral therapy-Naïve subjects with progressive HIV-1 subtype C infection. PloS One 8(6), e65950. Kourí, V., Alemán, Y., Díaz, D., Pérez, L., Limia, C.M., Soto, Y., Martínez, Y., Baños, Y., Caturla, Y., Campos, J., Ortega, L.M., Pérez, J. and Hengge U. 2016. Co-Receptor tropism determined by genotypic assay in HIV-1 circulating in Cuba. J. AIDS. Clin. Res. 7(592), 7. Kouri, V., Aleman, Y., Perez, L., Perez, J., Fonseca, C., Correa, C., Aragonés, C., Campos, J., Álvarez, D., Schrooten, Y., Dekeersmaeker, N., Imbrechts, S., Beheydt, G., Vinken, L., Pérez, D., Álvarez, A., Soto, Y., Vandamme, A.M. and Van Laethem, K. 2012. High frequency of antiviral drug resistance and non-B subtypes in HIV-1 patients failing antiviral therapy in Cuba. J. Clin. Virol. 55(4), 348–355. Kouri, V., Khouri, R., Aleman, Y., Abrahantes, Y., Vercauteren, J., Pineda-Pena, A.C., Theys, K., Megens, S., Moutschen, M., Pfeifer, N., Van Weyenbergh, J., Pérez, A.B., Pérez, J., Pérez, L., Van Laethem, K. and Vandamme, A.M. 2015. CRF19_cpx is an evolutionary fit HIV-1 variant strongly associated with rapid progression to AIDS in Cuba. EBioMedicine 2(3), 244–254. Kourí, V., Pérez, L., Alemán, Y., Pérez, J., Fonseca, C., Pintos, Y. 2019. Molecular epidemiology of clinical HIV-1 sequences isolated between 2008–2018 in Cuba. In 10th IAS Conference on HIV Science (IAS 2019), 2019 July. Lemoine, F., Correia, D., Lefort, V., Doppelt-Azeroual, O., Mareuil, F., Cohen-Boulakia, S. and Gascuel, O. 2019. NGPhylogeny.fr: new generation phylogenetic services for non-specialists. Nucleic. Acids. Res. 47, W260–W265. Machado, L.Y., Pintos, Y., Díaz, H.M., Pérez, L., Blanco, M., Kourí, V., Aleman, Y. and Martinez, L. 2017. Increase of recombinant forms CRF20, 23, 24_BG and several URF of HIV-1 among newly diagnosed Cuban patients: 2013-2014. ARC. J. AIDS. 2(1), 24–31. Najera, R., Delgado, E., Perez-Alvarez, L. and Thomson, M.M. 2002. Genetic recombination and its role in the development of the HIV-1 pandemic. AIDS (London, England) 16(Suppl 4), S3–S16. Oberle, C.S., Joos, B., Rusert, P., Campbell, N.K., Beauparlant, D., Kuster, H., Weber, J., Schenkel, C.D., Scherrer, A.U., Magnus, C., Kouyos, R., Rieder, P., Niederöst, B., Braun, D.L., Pavlovic, J., Böni, J., Yerly, S., Klimkait, T., Aubert, V., Trkola, A., Metzner, K.J. and Günthard, H.F. 2016. Tracing HIV-1 transmission: envelope traits of HIV-1 transmitter and recipient pairs. Retrovirology 13(1), 62. Palmer, J. and Poon, A.F.Y. 2019. Phylogenetic measures of indel rate variation among the HIV-1 group M subtypes. Virus. Evol. 5(2), vez022. Perez, L., Thomson, M.M., Bleda, M.J., Aragones, C., Gonzalez, Z., Perez, J., Sierra, M., Casado, G., Delgado, E. and Nájera, R. 2006. HIV type 1 molecular epidemiology in cuba: high genetic diversity, frequent mosaicism, and recent expansion of BG intersubtype recombinant forms. AIDS. Res. Hum. Retroviruses. 22(8), 724–733. Sede, M.M., Moretti, F.A., Laufer, N.L., Jones, L.R. and Quarleri, J.F. 2014. HIV-1 tropism dynamics and phylogenetic analysis from longitudinal ultra-deep sequencing data of CCR5- and CXCR4-using variants. PloS One 9(7), e102857. Sierra, S., Kaiser, R., Thielen, A. and Lengauer, T. 2007. Genotypic coreceptor analysis. Eur. J. Med. Res. 12(9), 453–462. Smyth, R.P., Davenport, M.P. and Mak, J. 2012. The origin of genetic diversity in HIV-1. Virus. Res. 169(2), 415–429. Utachee, P., Nakamura, S., Isarangkura-Na-Ayuthaya, P., Tokunaga, K., Sawanpanyalert, P., Ikuta, K., Auwanit, W. and Kameoka, M. 2010. Two N-linked glycosylation sites in the V2 and C2 regions of human immunodeficiency virus type 1 CRF01_AE envelope glycoprotein gp120 regulate viral neutralization susceptibility to the human monoclonal antibody specific for the CD4 binding domain. J. Virol. 84(9), 4311–4320. Vandekerckhove, L.P., Wensing, A.M., Kaiser, R., Brun-Vézinet, F., Clotet, B., De Luca, A., Dressler, S., Garcia, F., Geretti, A.M., Klimkait, T., Korn, K., Masquelier, B., Perno, C.F., Schapiro, J.M., Soriano, V., Sönnerborg, A., Vandamme, A.M., Verhofstede, C., Walter, H., Zazzi, M., Boucher, C.A. and European Consensus Group on clinical management of tropism testing. 2011. European guidelines on the clinical management of HIV-1 tropism testing. Lancet. Infect. Dis. 11(5), 394–407. Van Laethem, K., Schrooten, Y., Lemey, P., Van Wijngaerden, E., De Wit, S., Van Ranst, M. and Vandamme, A.M. 2005. A genotypic resistance assay for the detection of drug resistance in the human immunodeficiency virus type 1 envelope gene. J. Virol. Methods. 123(1), 25–34. Westby, M. 2007. Resistance to CCR5 antagonists. Curr. Opin. HIV. AIDS. 2(2), 137–144. Zhang, L., Ma, L., Wang, Z., Wang, Y., Zhang, J., Wang, H. and Shao, Y. 2014. Alterations in HIV-1 gp120 V3 region are necessary but not sufficient for coreceptor switching in CRF07_BC in China. PloS One 9(3), e93426. Zolla-Pazner, S. and Cardozo, T. 2010. Structure-function relationships of HIV-1 envelope sequence-variable regions refocus vaccine design. Nat. Rev. Immunol. 10(7), 527–535. | ||
| How to Cite this Article |
| Pubmed Style Batista AS, Santos LP, Zaldívar LYM, González MM, Saavedra YP, Ávila JP, Torres HMD, Núñez TR, Puertas JME, Morales YB, Romero EN, Cardellá VK. Tropism prediction in HIV-1 variants circulating in Cuba, 2017-2019: Implications for the pathogenesis of infection and response to Maraviroc. J Microbiol Infect Dis. 2023; 13(2): 59-71. doi:10.5455/JMID.2023.v13.i2.3 Web Style Batista AS, Santos LP, Zaldívar LYM, González MM, Saavedra YP, Ávila JP, Torres HMD, Núñez TR, Puertas JME, Morales YB, Romero EN, Cardellá VK. Tropism prediction in HIV-1 variants circulating in Cuba, 2017-2019: Implications for the pathogenesis of infection and response to Maraviroc. https://www.jmidonline.org/?mno=302657413 [Access: July 02, 2025]. doi:10.5455/JMID.2023.v13.i2.3 AMA (American Medical Association) Style Batista AS, Santos LP, Zaldívar LYM, González MM, Saavedra YP, Ávila JP, Torres HMD, Núñez TR, Puertas JME, Morales YB, Romero EN, Cardellá VK. Tropism prediction in HIV-1 variants circulating in Cuba, 2017-2019: Implications for the pathogenesis of infection and response to Maraviroc. J Microbiol Infect Dis. 2023; 13(2): 59-71. doi:10.5455/JMID.2023.v13.i2.3 Vancouver/ICMJE Style Batista AS, Santos LP, Zaldívar LYM, González MM, Saavedra YP, Ávila JP, Torres HMD, Núñez TR, Puertas JME, Morales YB, Romero EN, Cardellá VK. Tropism prediction in HIV-1 variants circulating in Cuba, 2017-2019: Implications for the pathogenesis of infection and response to Maraviroc. J Microbiol Infect Dis. (2023), [cited July 02, 2025]; 13(2): 59-71. doi:10.5455/JMID.2023.v13.i2.3 Harvard Style Batista, A. S., Santos, . L. P., Zaldívar, . L. Y. M., González, . M. M., Saavedra, . Y. P., Ávila, . J. P., Torres, . H. M. D., Núñez, . T. R., Puertas, . J. M. E., Morales, . Y. B., Romero, . E. N. & Cardellá, . V. K. (2023) Tropism prediction in HIV-1 variants circulating in Cuba, 2017-2019: Implications for the pathogenesis of infection and response to Maraviroc. J Microbiol Infect Dis, 13 (2), 59-71. doi:10.5455/JMID.2023.v13.i2.3 Turabian Style Batista, Anamary Suárez, Lissette Pérez Santos, Liuber Yans Machado Zaldívar, Melissa Méndez González, Yanet Pintos Saavedra, Jorge Pérez Ávila, Héctor Manuel Díaz Torres, Tamara Rojas Núñez, Juliet María Enriquez Puertas, Yoanna Baños Morales, Enrique Noa Romero, and Vivian Kourí Cardellá. 2023. Tropism prediction in HIV-1 variants circulating in Cuba, 2017-2019: Implications for the pathogenesis of infection and response to Maraviroc. Journal of Microbiology and Infectious Diseases, 13 (2), 59-71. doi:10.5455/JMID.2023.v13.i2.3 Chicago Style Batista, Anamary Suárez, Lissette Pérez Santos, Liuber Yans Machado Zaldívar, Melissa Méndez González, Yanet Pintos Saavedra, Jorge Pérez Ávila, Héctor Manuel Díaz Torres, Tamara Rojas Núñez, Juliet María Enriquez Puertas, Yoanna Baños Morales, Enrique Noa Romero, and Vivian Kourí Cardellá. "Tropism prediction in HIV-1 variants circulating in Cuba, 2017-2019: Implications for the pathogenesis of infection and response to Maraviroc." Journal of Microbiology and Infectious Diseases 13 (2023), 59-71. doi:10.5455/JMID.2023.v13.i2.3 MLA (The Modern Language Association) Style Batista, Anamary Suárez, Lissette Pérez Santos, Liuber Yans Machado Zaldívar, Melissa Méndez González, Yanet Pintos Saavedra, Jorge Pérez Ávila, Héctor Manuel Díaz Torres, Tamara Rojas Núñez, Juliet María Enriquez Puertas, Yoanna Baños Morales, Enrique Noa Romero, and Vivian Kourí Cardellá. "Tropism prediction in HIV-1 variants circulating in Cuba, 2017-2019: Implications for the pathogenesis of infection and response to Maraviroc." Journal of Microbiology and Infectious Diseases 13.2 (2023), 59-71. Print. doi:10.5455/JMID.2023.v13.i2.3 APA (American Psychological Association) Style Batista, A. S., Santos, . L. P., Zaldívar, . L. Y. M., González, . M. M., Saavedra, . Y. P., Ávila, . J. P., Torres, . H. M. D., Núñez, . T. R., Puertas, . J. M. E., Morales, . Y. B., Romero, . E. N. & Cardellá, . V. K. (2023) Tropism prediction in HIV-1 variants circulating in Cuba, 2017-2019: Implications for the pathogenesis of infection and response to Maraviroc. Journal of Microbiology and Infectious Diseases, 13 (2), 59-71. doi:10.5455/JMID.2023.v13.i2.3 |

